Favorable clinical course after early-intensive immunotherapy for new-onset refractory status epilepticus
Article information

Abstract
Background
New-onset refractory status epilepticus (NORSE) refers to the newly established concept of a disease characterized by refractory status epilepticus without an identifiable etiology. Recent reports have indicated the importance of immunotherapy for NORSE.
Case Report
A 37-year-old man with no past history of epilepsy was admitted for a presenting complaint of confusion. He was treated with acyclovir and anti-epileptic drug (AED) for presumed herpes encephalitis. However, he developed generalized tonic-clonic seizures on day 6 of admission that worsened despite treatment with multiple AEDs. NORSE was considered to be a probable diagnosis and immunotherapy with methylprednisolone and immunoglobulin was scheduled. However, persistent seizure activity was observed on the electroencephalogram after the completion of initial immunotherapy. Subsequently, rituximab was administered for 4 weeks. He eventually regained consciousness and was able to resume social activity.
Conclusion
Our patient exhibited a favorable outcome with early-intensive immunotherapy and subsequent rituximab treatment for NORSE.
INTRODUCTION
Although there is lack of consensus regarding the definition of refractory status epilepticus (RSE), it can be defined as continuous or repetitive seizures that do not respond to first and second-line antiepileptic drug (AED) therapy [1]. RSE is considered to be a life-threatening emergency with a high mortality rate of at least 15% or even 40%, if it is left untreated [2].
Currently, immunotherapy is thought to be an important newly established concept for treating new-onset refractory status epilepticus (NORSE), which is characterized by RSE, without an identifiable etiology in otherwise healthy individuals [3].
We describe a favorable outcome in a patient with NORSE, who was treated with early-intensive immunotherapy in the form of sequential administration of intravenous methylprednisolone and immunoglobulin, followed by subsequent rituximab administration within 2 weeks of initiating immunotherapy.
CASE REPORT
A 37-year-old man with no past history of epilepsy was transferred to our center. He presented with confusion preceded by fever, chills, and headache. Baseline laboratory tests and computed tomography imaging revealed no definite evidence of systemic inflammation/infection. Cerebrospinal fluid (CSF) examination was unremarkable, except for a mild elevation in CSF proteins (60.7 mg/dL), with an opening pressure of 16 cm, no cells (0/μL), and a glucose level of 95 mg/dL (for comparison, the serum glucose level was 150 mg/dL). Red blood cells were not detected on CSF. Staining, culture, antibodies, and polymerase chain reaction tests for the detection of bacteria, fungi, viruses, and mycobacterium tuberculosis in the CSF were negative. CSF cytology for the detection of abnormal malignant cells was also negative. Serum influenza virus A/B antigen, herpes simplex virus, and varicella-zoster virus antibodies, blood culture, and autoimmune antibodies (e.g., antinuclear, antidouble-stranded DNA, antithyroid peroxidase, antithyroglobulin, and antineutrophilic cytoplasmic antibodies) were absent. Brain magnetic resonance imaging (MRI) revealed a diffusion restricted lesion in the right medial temporal lobe with increased gadolinium enhancement (Fig. 1A, red line). Baseline electroencephalogram (EEG) (obtained at admission) revealed slow background activity.
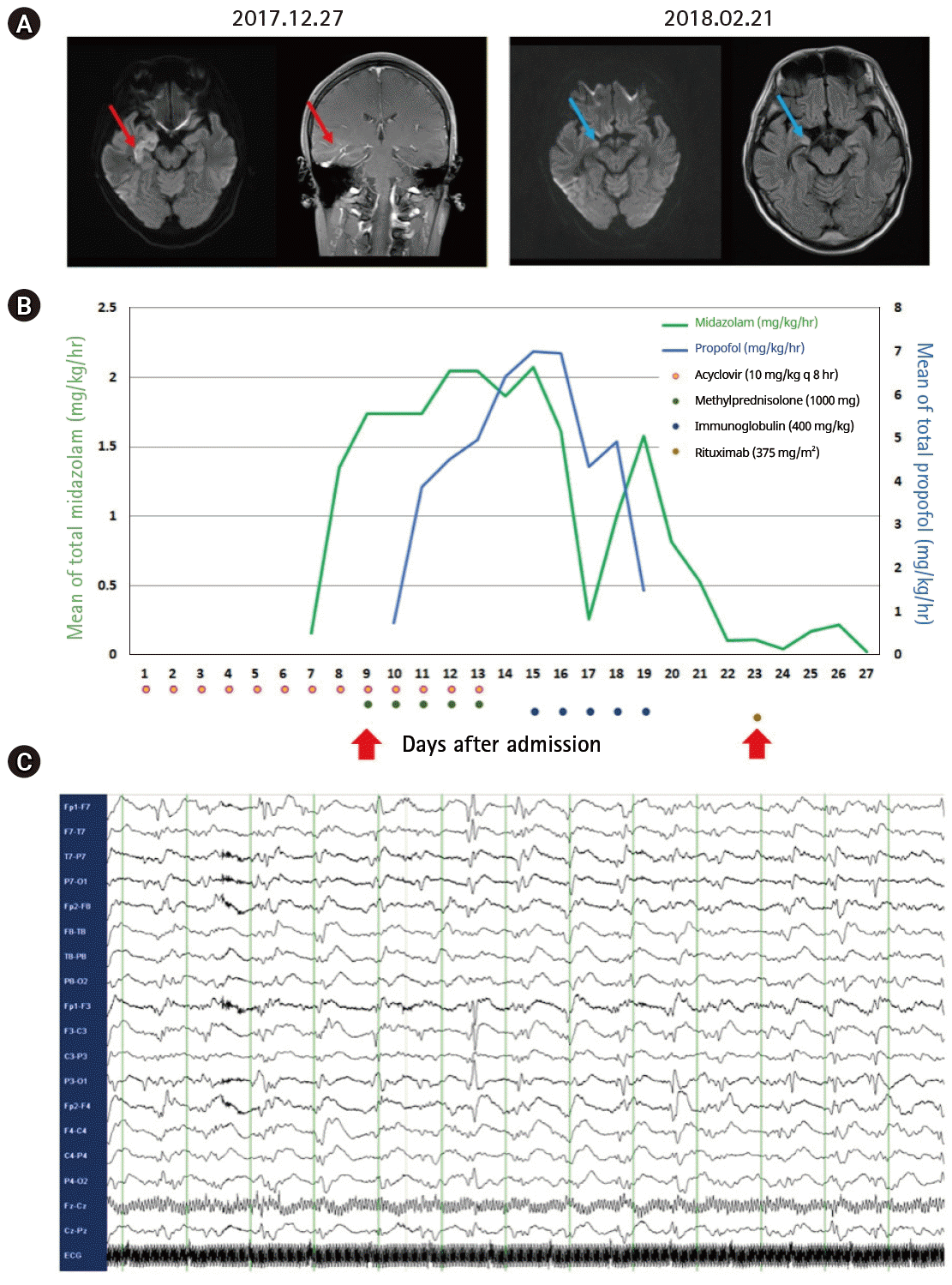
(A) Follow-up brain magnetic resonance imaging showing disappearance of the diffusion-restricted lesion with remnant fluid-attenuated inversion recovery changes in the right medial temporal lobe (blue lines), which previously showed an increased diffusion restriction and gadolinium enhancement (red lines). (B) Continuous infusion of multiple anesthetics including midazolam and propofol was increased sequentially for the management of refractory status epilepticus, but were tapered during sequential immunotherapy (methylprednisolone 1,000 mg for 5 days and intravenous immunoglobulin [IVIG] 0.4 g/kg for 5 days), followed by the first infusion of rituximab 375 mg/m2 as booster immunotherapy (red arrow), after the maximum possible administration of multiple antiepileptic drugs including levetiracetam 1,500 mg, every 12 hours; valproic acid 800 mg, every 6 hours; phenytoin 300 mg, every 8 hours; phenobarbital 200 mg, every 8 hours; and topiramate 200 mg, every 12 hours. (C) Rituximab was started on day 23 of hospitalization, based on the findings of bilateral periodic lateralized epileptiform discharge on continuous electroencephalography monitoring even 3 days after the completion of initial immunotherapy, with a regimen of once a week for 4 weeks.
The patient was administered acyclovir 10 mg/kg, every 8 hours, and levetiracetam 500 mg, every 12 hours, following a probable diagnosis of herpes encephalitis. However, episodes of focal seizures with lip smacking and eyeball deviation to the left side increased in frequency, which were accompanied by a newly developed epileptiform discharge on the EEG that required multiple AEDs: valproic acid 600 mg, every 8 hours; phenytoin 200 mg, every 8 hours; levetiracetam 1,000 mg, every 12 hours; and phenobarbital 100 mg, every 8 hours.
On day 6, he developed a generalized tonic-clonic seizure, which lasted for more than 5 minutes, which worsened, and led to a drastic increase in the need for multiple anesthetics and AEDs. A probable diagnosis of NORSE was made at this stage and immunotherapy was scheduled (Fig. 1B). Intravenous methylprednisolone and immunoglobulin were sequentially administered, which alleviated the seizure activity, thereby reducing the need for anesthetics. However, persistent seizure activity was noted on the EEG 3 days after the completion of intravenous methylprednisolone and immunoglobulin treatment (Fig. 1C). Rituximab was administered, and the seizure activity resolved completely on EEG 4 days after the first infusion, prompting discontinuation of anesthetic infusion. He regained consciousness and could follow instructions 4 days after the discontinuation of anesthetics.
At this stage, the test results for the detection of autoimmune encephalitis antibodies (e.g., NMDAR, AMPA1, AMPA2, LGI1, CASPR2, and GABA-B) and paraneoplastic antibodies (e.g., Hu, Yo, Ri, Ma2, CV2/CRMP5, amphiphysin, recoverin, SOX1, and titin) in serum and CSF were negative. Follow-up brain MRI revealed an improvement in what was probably herpes encephalitis or limbic encephalitis compared to the earlier lesion on the baseline image (Fig. 1A, blue line). He completed a 4-week schedule of rituximab treatment and successfully returned to social activity after 2 months of hospitalization.
DISCUSSION
We described the favorable outcome of early-intensive immunotherapy for a 37-year-old man with RSE. Treatment was initiated immediately on suspicion of NORSE with a 5-day course of intravenous methylprednisolone and immunoglobulin, followed by the first infusion of rituximab within 2 weeks of immunotherapy. This drug regimen kept the patient seizure-free for a period of 1-month after hospitalization.
Although NORSE was initially defined as cryptogenic RSE, recent studies have suggested that autoimmune encephalitis may be a common etiology [4,5]. Therefore, immunotherapy has been recommended for NORSE, even in the absence of the detection of specific antibodies, after excluding infections from the differential diagnosis [6]. Furthermore, several recent systematic reviews have reported that early treatment is associated with improved outcomes in autoimmune encephalitis [5]. Therefore, earlier empirical immunotherapies such as steroid-pulse therapy, immunoglobulins, or plasmapheresis are often tried sequentially, depending on the clinical response.
Interestingly, although our patient exhibited improvement in the clinical course during initial immunotherapy (e.g., sequential treatment with intravenous methylprednisolone and immunoglobulin), which contributed to a dramatic decrease in the need for anesthetics, total discontinuation of anesthetics was possible with the subsequent administration of rituximab (for the management of RSE). Rituximab is a monoclonal antibody against CD20-positive B cells. It leads to B-cell depletion and therefore, suppression of autoimmune neurological disorders including autoimmune encephalitis, especially in patients that respond poorly or are unresponsive to empirical immunotherapy [7,8]. Rituximab does not affect the innate immune system, immunoglobulin, or T-cell activity, and is relatively safe in terms of infectious adverse events [9]. Therefore, rituximab can be considered to be an early immune booster for patients with NORSE, who are resistant to empirical immunotherapy, with or without proven autoantibodies. Prolonged seizure activity can aggravate epileptogenicity, which in turn leads to an increased risk of super-RSE by a kindling mechanism [10].
Notes
Conflict of interest
No potential conflict of interest relevant to this article.
Funding
This work was supported by a 2-year Research Grant of Pusan National University.
Author contributions
Conceptualization: HSK, SHA. Visualization & Writing–original draft: HSK, JK, BJH, KNW, MGP, KPP, SHA. Writing–review editing: HSK, JK, BJH, KNW, MGP, KPP, SHA.