Brain injury in extracorporeal cardiopulmonary resuscitation: translational to clinical research
Article information

Abstract
The addition of extracorporeal membrane oxygenation (ECMO) to conventional cardiopulmonary resuscitation (CPR), termed extracorporeal cardiopulmonary resuscitation (ECPR), has significantly improved survival in selected patient populations. Despite this advancement, significant neurological impairment persists in approximately half of survivors. ECPR represents a potential advancement for patients who experience refractory cardiac arrest (CA) due to a reversible etiology and do not regain spontaneous circulation. Important risk factors for acute brain injury (ABI) in ECPR include lack of perfusion, reperfusion, and altered cerebral autoregulation. The initial hypoxic-ischemic injury caused by no-flow and low-flow states after CA and during CPR is compounded by reperfusion, hyperoxia during ECMO support, and nonpulsatile blood flow. Additionally, ECPR patients are at risk for Harlequin syndrome with peripheral cannulation, which can lead to preferential perfusion of cerebral vessels with deoxygenated blood. Lastly, the oxygenator membrane is prothrombotic and requires systemic anticoagulation. The two competing phenomena result in thrombus formation, hemolysis, and thrombocytopenia, increasing the risk of ischemic and hemorrhagic ABI. In addition to clinical studies, we assessed available ECPR animal models to identify the mechanisms underlying ABI at the cellular level. Standardized multimodal neurological monitoring may facilitate early detection of and intervention for ABI. With the increasing use of ECPR, it is critical to understand the pathophysiology of ABI, its prevention, and the management strategies for improving the outcomes of ECPR. Translational and clinical research focusing on acute ABI immediately after ECMO cannulation and its short- and long-term neurological outcomes are warranted.
INTRODUCTION
The use of extracorporeal membrane oxygenation (ECMO) to treat patients with cardiac arrest (CA) was first described in 1957 when several patients with CA refractory to conventional cardiopulmonary resuscitation (CCPR) including open cardiac massage were placed on cardiopulmonary bypass (CPB), allowing time for an attempt at definitive management [1]. A percutaneous system for ECMO cannulation initiated at the bedside was successfully implanted in five patients in 1983 by Phillips et al. [2]. Percutaneous ECMO was successfully applied to CA patients refractory to CCPR, which was termed extracorporeal cardiopulmonary resuscitation (ECPR), in 1991 when a specialized team achieved the 6-month survival of 64% of adult patients (n=11) who had refractory CA close to a cardiac operating room [3]. A similar survival pattern was reported for children in 1992 when 11 patients aged 8–22 months were treated with ECMO for CA refractory to CCPR [4]. With the development of more portable ECMO devices and percutaneous implantation, ECPR has become possible in medical intensive care units and emergency rooms, expanding its potential use in patients with out-of-hospital cardiac arrest (OHCA) [5,6]. Multiple animal models have demonstrated improved survival and end-organ protection with ECPR, compared with CCPR, after CA [7-9]
Since the mid-2000s, the number of ECMO-capable centers participating in the Extracorporeal Life Support Organization (ELSO) has tripled, increasing the feasibility of ECPR [10-12]. Two meta-analyses of observational studies comparing ECPR and CCPR for patients with CA occurring in and outside of the hospital demonstrated an improvement in both survival and neurologic outcomes with ECPR. Both analyses showed improved survival among patients with in-hospital cardiac arrest (IHCA) on discharge and at 1-year follow-up. Patients with OHCA showed no difference in survival or neurologic outcomes on discharge; however, after 3–6 months, improvements in survival and neurologic outcomes were observed [13,14]. ECPR continues to be an active area of innovation; several clinical trials of emergency medical response teams, including providers capable of ECMO cannulation in the field as an effort to improve the ECPR outcomes, have been conducted [15].
CA INCIDENCE AND SURVIVAL
In the United States, the estimated annual incidences of IHCA and OHCA are 300,000 and 350,000–400,000, respectively. Globally, measures of incidence are lacking and vary significantly due to regional differences, for example, between developed and developing nations and rural and urban areas, with variable recordings of CA and outcomes. Multiple regional efforts are underway to determine the incidence and standardize the reporting of CA to more accurately measure survival and other outcomes [16-26]. In the United States, up to 25% of cases survive up to hospital discharge after IHCA, with the majority having favorable neurologic recovery demonstrated by a cerebral performance score (CPC) of 1 or 2 [14,16,27]. However, a recent meta-analysis reported only a 13% 1-year survival after IHCA [28]. OHCA survival is difficult to estimate because several patients are deceased before transport to the hospital, limiting the accurate recording of OHCA numbers. A review of emergency medical services (EMS) records indicates that approximately 6%–10% of patients who visit the hospital survive up to discharge [16-21].
ECPR INDICATIONS AND ELIGIBILITY
Although the optimal application of ECPR has not been established, the published literature has similar inclusion and exclusion criteria with limited data. The implementation of ECPR requires the rapid assembly and coordination of a specialized team, including members capable of cannulating ECMO, a perfusionist or specialist to monitor the ECMO circuit and flows, and trained nursing support. Appropriate patient selection relies on timely assessments and effective communication from the resuscitation team, including appropriate identification of ECPR-eligible patients, as well as determining the timing for transition to ECMO cannulation and transport during cardiopulmonary resuscitation (CPR).
For eligibility, there is a paucity of data to support age cutoffs in selecting ECMO candidates. Age of <70 years is recommended by the ELSO based on retrospective data and <75 years by some experienced regional centers with OHCA ECPR protocols [29-33]. Patients should have a minimum no-flow time, defined as the time between CA and the initiation of CPR. The goal is to have a no-flow time of <5 minutes, which may depend on the presence of a witnessed CA and bystander CPR before the arrival of EMS for OHCA. The target time from CA to the initiation of ECMO (low-flow time) was <60 minutes [30].
An initial rhythm of ventricular fibrillation (VF) or ventricular tachycardia (VT) is suggestive of a primary reversible cardiac etiology for CA, and OHCA patients can be considered for early transportation to an ECPR-capable center if VT/VF is refractory after three shocks [30,31,33]. This strategy allows early identification and transport of patients to an ECPR-capable facility with continued CPR and pre-notification to the cardiac catheterization laboratory to prepare for immediate ECMO cannulation and cardiac catheterization. For patients with IHCA, ECMO cannulation can be considered after CA refractory to 10–20 minutes of CCPR if there is a suspected reversible etiology or after three shocks for VT/VF. Markers of perfusion that may aid in patient selection include those with end-tidal CO2 of >10 mmHg measured during CPR by capnography, PaO2 of >55 mmHg (O2 saturation >85%), and lactate of <18 mmol/L. However, they are often not available before cannulation, and there are limited data on their association with outcomes [15,31,34,35]. The exclusion criteria included significant comorbidities such as terminal disease, advanced cancer, advanced neurological disease, and low-performance status before CA [30,31].
The optimal timing for the transition from CCPR to ECPR has not yet been established. Careful consideration and further research are necessary to determine the ideal ECPR eligibility and timing to improve outcomes and minimize unnecessary ECMO cannulation. A retrospective analysis of OHCA demonstrated benefits related to survival and neurologic outcomes measured by CPC at 3 months beginning after CPR for 21 minutes with propensity-matched patients having improved outcomes if they were treated with ECPR, compared with CCPR, if the duration was >21 minutes and no survival benefit if initiated earlier [36]. As the use of ECPR increases, early detection of acute brain injury (ABI) and the improvement of neurological outcomes are crucial in improving overall outcomes in this population. Fig. 1 outlines the proposed mechanisms of ABI in patients after ECPR.
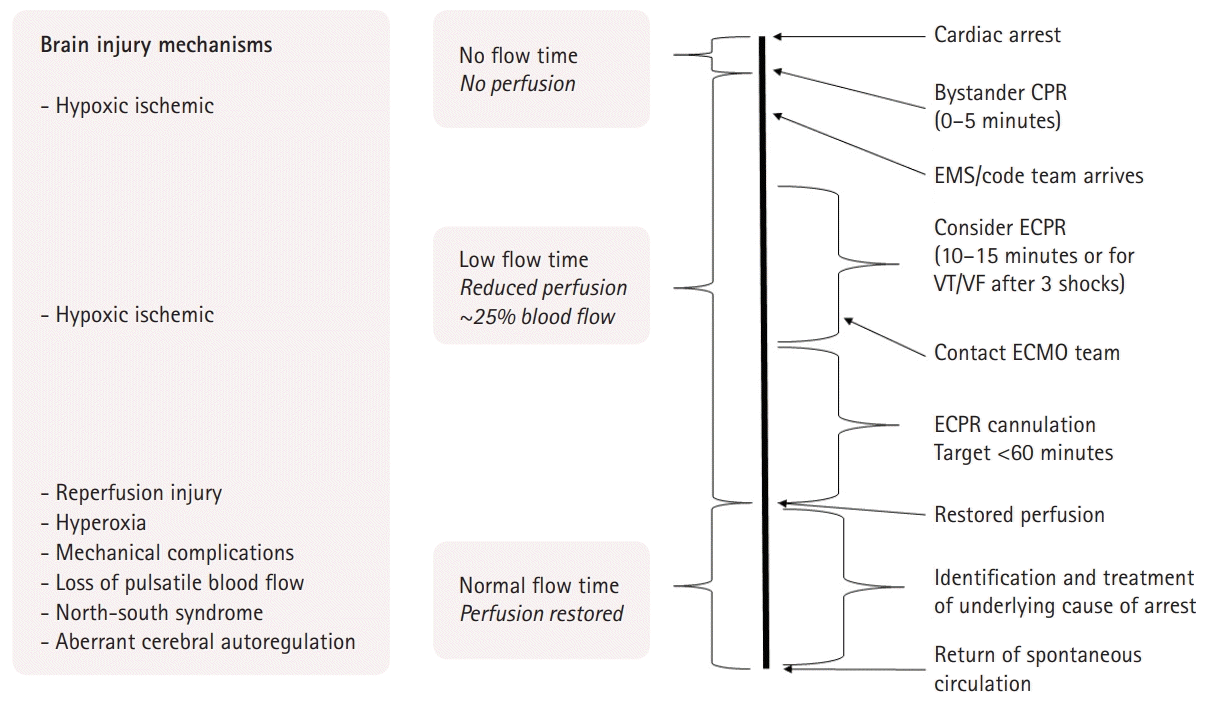
Extracorporeal cardiopulmonary resuscitation (ECPR) model with associated risk factors for brain injury. This figure represents the model for the proposed timing for ECPR. ECPR should be considered if return of spontaneous circulation (ROSC) is not obtained within 10–15 minutes or after 3 shocks for ventricular tachycardia (VT)/ventricular fibrillation (VF). The cannulation goal is <60 minutes. Perfusion is restored after cannulation; however, ROSC may not be achieved until the underlying cause is addressed. The left column shows proposals of brain injury mechanisms during different stages of resuscitation. Flow time refers to duration in minutes. Bystander cardiopulmonary resuscitation (CPR) refers to life support measures initiated by on-scene persons before the arrival of emergency medical services (EMS) or health care agents before the arrival of the code team. ECMO, extracorporeal membrane oxygenation.
SOCIETY GUIDELINES ON ECPR
In 2020, the American Heart Association stated that there was insufficient evidence in support of the routine use of ECPR, but the therapy may be considered in select patients with suspected reversible etiology of CA [37]. In 2021, the European Resuscitation Council weakly recommended the consideration of ECPR for cases of refractory CA because of the low level of evidence [38]. None of the organizations provided specific indications for ECPR. The ELSO recently published a consensus statement for ECPR. Although ELSO did not provide specific guidelines or inclusion criteria due to the lack of strong evidence, they highlighted the importance of appropriately trained healthcare providers, teamwork, and planning. The ELSO recommended regional inclusion criteria, including resource availability and capability, to maximize favorable neurologic outcomes. They provided sample inclusion criteria, which included age of <70 years, witnessed CA, arrest to CPR (no-flow time) of <5 minutes, initial rhythm VT/VF, pulseless electrical activity for IHCA, end-tidal CO2 of >10 mmHg during CPR, absence of life-limiting comorbidity, and absence of known aortic valve incompetence [12,30]. The current guidelines do not provide standard neurological definitions and monitoring and management recommendations.
SURVIVAL AND OUTCOMES AFTER ECPR
Survival of up to 30% of appropriately selected patients after CA refractory to CCPR treated with ECPR has been reported [12,39]. However, there is significant variability in survival based on the location of CA, with one study reporting 50% survival if CA occurred in or near a cardiac catheterization lab and 15% for other locations [11]. A recent randomized controlled trial, the Advanced reperfusion strategies for patients with out-of-hospital cardiac arrest and refractory ventricular fibrillation (ARREST): a phase 2, single centre, open-label, randomized controlled trial, compared ECPR and CCPR for OHCA with VF or VT rhythms persisting after three defibrillations. The trial was discontinued early with a significant survival benefit of ECPR over CCPR (43% vs. 7%) [30]. The ARREST trial demonstrated that a streamlined systematic approach in a high-volume ECMO center with experienced staff could dramatically improve the ECPR outcomes; however, the results of this study lack generalizability and should be validated in other centers with larger samples [40].
Among survivors of CA, long-term neurological sequelae, such as cognitive impairment and difficulties in performing activities of daily life, are common. The prevalence of hypoxic-ischemic brain injuries (HIBIs) in OHCA is higher with CCPR than with ECPR among survivors (50% vs. 23%) [41,42]. Although several of these survivors continue to improve toward near or complete functional independence in the long term, they still have a poor quality of life [43-45]. Despite the survival benefits and possibly improved neurological outcomes with ECPR, compared with CCPR, there are sparse data on the long-term function and quality of life of ECPR survivors [13,14,40]. As the data on ABI after ECPR accumulate, it is important to study the long-term neurological outcomes in this population.
RANDOMIZED CLINICAL TRIALS
The ARREST trial was a phase 2 open-label randomized clinical trial (RCT) of ECPR compared with CCPR in OHCA patients who presented initially with VF or VT. Participants were eligible if their dysrhythmia was refractory to three defibrillation shocks and were randomized to continue CCPR or ECPR. The trial was discontinued early after the pre-specified analysis showed clear superiority in the ECPR arm. The survival rate was 43% at discharge, and all patients were alive at 6 months with favorable CPC scores, with medians of 2.5 at discharge and 1.2 at 6 months, respectively, in the patients receiving ECPR. In contrast, patients in the CCPR group had a 0% survival within 6 months [40]. Another RCT, the Prague OHCA study (NCT 01511666), compared the standard care of CPR in the field with immediate transport using CPR assisted with a mechanical device for chest compressions followed by ECPR upon arrival at the hospital if return of spontaneous circulation (ROSC) was not obtained. Survival at 180 days was not significantly different (CCPR, 22% vs. ECPR, 31.5%); however, neurologic recovery at 30 days was significantly improved in the ECPR group (22.7% vs. 34.7%) as was survival in the group with prolonged CA (>45 minutes) receiving CPR [46]. Despite these encouraging outcomes, these findings need to be replicated with multicenter RCTs to validate the findings and possibly generalize to different populations. Other ongoing RCTs evaluating ECPR at the time of this article include the APACAR2 trial (NCT02527031), the Extracorporeal Cardiopulmonary Resuscitation for Refractory Out-of-Hospital Cardiac Arrest (EROCA): Results of a Randomized Feasibility Trial of Expedited Out-of-Hospital Transport (NCT03065647), and the Early Initiation of Extracorporeal Life Support in Refractory OHCA (INCEPTION) trial (NCT03101787) [47].
ABI IN ECPR: MECHANISTIC CONSIDERATION
Hypoxic-ischemic injury and reperfusion injury
HIBIs from the cessation of blood flow after CA is a leading cause of morbidity and mortality in CA survivors and has been reported in 23% of patients treated with ECPR [42,48,49]. This primary injury is followed by secondary ABI after ROSC because adequate blood supply is restored, leading to the formation of reactive oxygen species (ROS), alteration in microvasculature blood flow, and reperfusion injury [48,50-52]. These insults may be compounded by prolonged resuscitation and immediate restoration of oxygenated cerebral blood flow during ECPR.
The abatement of these injuries with targeted temperature management (TTM) by induction of mild hypothermia (32°C–34°C) in survivors of CA has been shown in multiple RCTs to improve survival and neurologic outcomes [53,54]. The ECMO circuit allows for rapid cooling for TTM, and the feasibility of cooling within minutes has been demonstrated in an adult swine model [55,56]. Survival benefits were demonstrated for TTM after ECPR; however, this was a single-center observational study, and other studies have found no benefit. Overall, there are no high-quality data on the impact of TTM on ECPR outcomes [53,54,57]. An additional consideration for TTM in this population is that ECMO-associated coagulopathy may be exacerbated by lower temperature targets and the need for systemic anticoagulation.
Retrospective studies of increased mean arterial pressure (MAP; >80 mmHg) after CA have demonstrated improved survival and neurologic outcomes after CA. However, clear benefits have not been found in RCTs, and the most recent American Heart Association guidelines state that targeting MAP of >80 mmHg may be beneficial, but data are lacking [37,58,59]. In the setting of ECPR, the addition of anticoagulation to prevent circuit clotting increases the risk of hemorrhage, especially in patients with a significant burden of HIBIs with fresh cerebral infarcts. Therefore, optimal blood pressure management is unknown and MAP of >65 mmHg may be recommended to ensure adequate cerebral perfusion without a significantly increased risk of intracerebral hemorrhage and minimize the increased afterload created by ECMO flow against native heart blood flow.
Hyperoxia and ROS
Oxygen therapy plays a paramount role in the success of CPR after CA. However, excessive oxygenation or hyperoxia (commonly defined as mild PaO2 >100 or 120 mmHg and severe PaO2 >300 mmHg) after ROSC can lead to ABI. The detrimental effect of hyperoxia on ABI has been demonstrated in several different diseases, including traumatic brain injury, ischemic and hemorrhagic stroke, aneurysmal subarachnoid hemorrhage, and HIBIs. The underlying mechanism of ABI is an insult by ROS to the lipid membrane, deoxyribonucleic acid, and proteins.
Additionally, in CA, prolonged periods of ischemia deplete cells of adenosine triphosphate (ATP), preventing the recycling of reducing agents that neutralize ROS [60]. A decrease in the production of ATP also disrupts Na+/K+ ATPase, which is responsible for maintaining membrane potential in neurons and leads to a Ca2+ influx that causes the release of cytochrome c, leading to neuronal cell death via apoptosis. During ECPR, ECMO provides immediate cerebral blood flow restoration, which may exacerbate reperfusion injury in patients who are vulnerable to HIBIs with a global cerebral ischemic insult. Furthermore, ECMO decannulation is known to induce a systemic inflammatory response syndrome response, leading to further production of ROS and compounding the potential for secondary ABI [61].
Animal CA and ECMO models on “hyperoxia”
Established canine models of VF-induced CA lasting for 10 minutes followed by ROSC, were divided into normoxia (PaO2, 80–120 mmHg) and hyperoxia (PaO2 >120 mmHg) groups. ROS formation increased in a dose-dependent manner with an increase in the PaO2 level. This correlated with increased disruption of the mitochondrial pyruvate dehydrogenase complex, an enzyme that produces reducing agents to nullify ROS in hyperoxia compared with normoxia groups. Postmortem examination demonstrated that the cerebral cortex and hippocampal neurons had increased disruption of pyruvate dehydrogenase complex and cell death specifically in neuronal Purkinje cells. Although both normoxia and hyperoxia increased inflammatory activation of microglial cells and macrophages, Purkinje cell loss was greater in the hyperoxia group [62]. Similar results were observed in a rat model of global cerebral ischemia, where normoxia was compared with hyperoxia after 10 minutes of bilateral carotid occlusion. At 7 and 30 days postintervention, more hippocampal neurons remained normal on histological examination in normoxic rats and hyperoxic rats [63].
In an ECMO model of global hypoxia, adult New Zealand White rabbits were cannulated with veno-venous (VV)- and veno-arterial (VA)-ECMO and subsequently hypoventilated to a PaO2 of 27 mmHg and a pH of <7.0 and injected with bacterial endotoxin. The ECMO circuit was utilized to reoxygenate after the hypoxic event with 100% sweep oxygen compared with the control, which was reoxygenated through ventilation. Both VV and VA-ECMO groups demonstrated significantly increased concentrations of malondialdehyde, a byproduct of lipid damage by ROS and a marker of oxidative injury, both in the lung tissue and plasma compared with the control. This suggests that reperfusion via the ECMO circuit after a global hypoxic event increases ROS and lipid membrane peroxidation, which can lead to alveolar damage [64,65]. Table 1 summarizes the established animal models of ECPR.
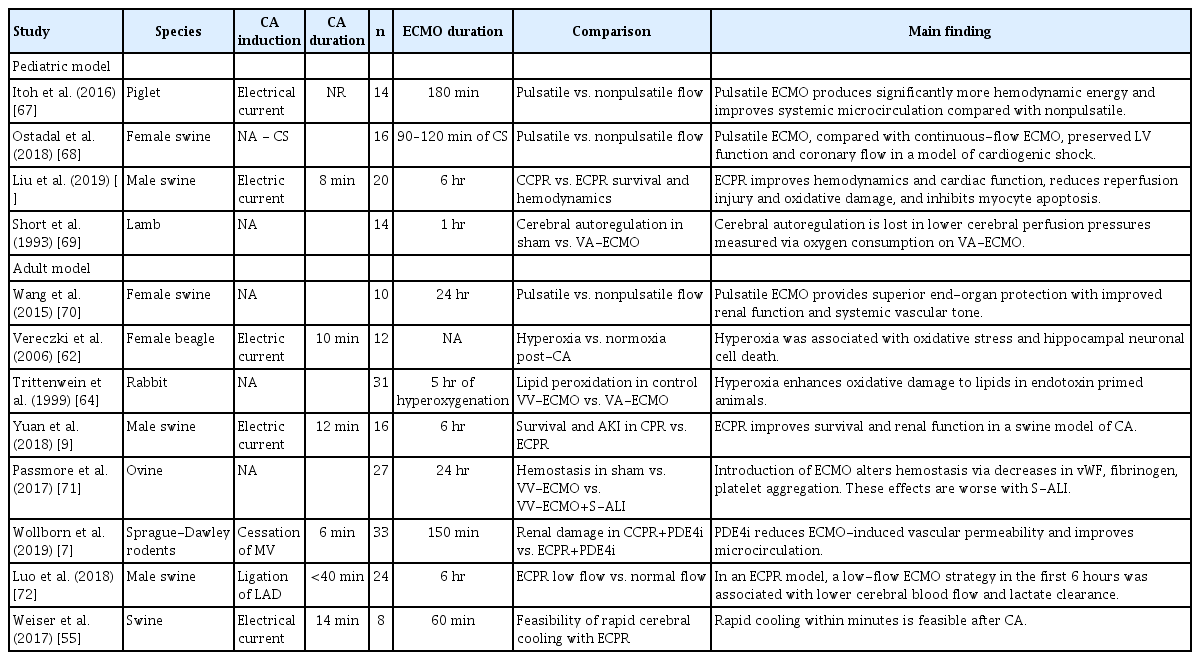
Comparison of animal models for CA, ECMO, and ECPR
Clinical research on “hyperoxia” in ECPR
Recently, the Conservative Oxygen Therapy during Mechanical Ventilation in the intensive care unit (ICU-ROX) study aimed to determine the benefits of conservative oxygen treatment and compare them with usual oxygen treatment for any mechanically ventilated patient in need of >24 hours of mechanical ventilation in the ICU [66]. The study compared the goal-directed oxygen treatment (FiO2 decreased to 0.21 as soon as possible with an upper limit alarm set for SpO2 ≥97%) to the usual oxygen treatment (standard care with no specific measures limiting FiO2 or SpO2) and found no difference in the number of ventilator-free days. However, in the subgroup analysis of patients with suspected HIBIs, mortality differed significantly between the goal-directed (conservative) oxygen group and the usual (standard) oxygen group (43% vs. 59%; reparatory rate, 0.73; 95% confidence interval, 0.54–0.99), highlighting the potential benefits of avoiding hyperoxia in patients at risk of brain ischemia. However, the effect of hyperoxia on ECPR patients remains unclear. In pediatric studies that have examined this specific question, moderate hyperoxia during the first 48 hours was an independent risk factor for increased mortality [73]. In a prior analysis of the ELSO registry from 2010 through 2015, hyperoxia, defined as a PaO2>100 mmHg at 24 hours, was associated with increased mortality in patients treated with VA-ECMO [74]. A second prospective single-center analysis identified that hyperoxia (PO2 >300 mmHg) during the first hour after resuscitation was associated with a worse neurologic outcome at discharge [75]. A recent analysis of 10,342 VA-ECMO patients from the ELSO showed that all subtypes of ABI were more common in patients with hyperoxia, as measured by 24-hour ABG [76]. However, data on this for ECPR patients are limited.
Animal models of nonpulsatile blood flow and cerebral autoregulation in ECMO
The loss of pulsatile blood flow occurs as a consequence of continuous flow ECMO systems and has been linked to endothelial dysfunction, increased sympathetic tone, decreased local oxygen consumption, and increased systemic vascular resistance [77-79]. An adolescent swine cardiogenic shock model was placed on VA-ECMO with a pulsatile (utilizing an electrocardiogram synchronized system) or nonpulsatile circuit, and cardiogenic shock was induced via balloon occlusion of the left main coronary artery [70,80]. ECMO flow was increased in a stepwise manner with intravascular measurement catheters showing significantly higher cardiac output, coronary artery blood flow, and MAP in patients with pulsatile blood flow than in those with nonpulsatile flow [68].
In a cerebral autoregulation model, newborn lambs were separated into the VA-ECMO group (n=7) or controls with right carotid artery and jugular vein ligation (n=7) and had an intracranial catheter inserted to manipulate cerebral perfusion pressure (CPP). Cerebral blood flow was maintained at lower CPPs in the control group, suggesting a loss of protective antiregulatory mechanisms in animals treated with VA-ECMO [69].
Loss of pulsatile flow and autoregulation in cardiac surgery
Pulsatile and nonpulsatile CPB flows were compared in 32 adult patients undergoing open-heart surgery with CPB. Cerebral blood flow velocity was measured using stereotactic transcranial Doppler (TCD) monitoring of the middle cerebral artery. Normal vasodilatory and constrictive responses to CO2 in the nonpulsatile group were blunted compared to the pulsatile flow, suggesting a loss of cerebral autoregulation in response to changes in CO2 with the loss of pulsatile blood flow [81]. In patients placed on CPB for surgery, the microcirculation of sublingual tissue was compared with pulsatile and nonpulsatile flow using a previously established noninvasive spectral imaging technique [82].
At the microcirculation level, disrupted perfusion and leukocyte activation in sublingual tissue were observed in the nonpulsatile group, compared with the pulsatile group. This effect was statistically significant, and it increased in magnitude with CPB time and was associated with lower lactate in the pulsatile flow group, suggesting an aberrant antiregulatory response with the loss of pulsatile flow [78]. In patients with implanted left ventricular assist devices, pulsatile flow devices (vs. nonpulsatile devices) showed significant reductions for stroke (9.9% vs. 19.4%) and disabling ischemic stroke (3.9% vs. 5.9%) two years post-implantation [83]. Non-pulsatile blood flow may impair cerebral autoregulation, reducing the normal protective response of cerebral vessels to changes in CPP and CO2 and further increasing the risk of ABI after CA; the feasibility and benefit of pulsatile flow during VA-ECMO and ECPR have yet to be established, but it warrants consideration as a mechanism for reducing ABI.
Animal model of impaired coagulation in ECMO
One animal model examined coagulopathy in adult sheep exposed to smoke-induced lung injury by comparing animals placed on VV-ECMO to controls (mechanical ventilation only). During 24 hours of ECMO flow, there was an increase in collagen-induced platelet aggregation, increased platelet aggregation time, and decreased clot firmness as measured by thromboelastography. Additionally, fibrinogen, factor VIII, and von Willebrand factor were all reduced in animals treated with VV-ECMO and were synergistically worse when combined with smoke-induced lung injury than in controls. This model suggests that abnormal platelet aggregation and decreased clot effectiveness in ECMO may predispose patients to coagulopathy, increasing the risk of thrombosis and bleeding [71].
Ischemic stroke and intracranial hemorrhage in VA-ECMO
In addition to HIBIs, acute ischemic stroke (AIS) and intracranial hemorrhage (ICH) are major complications that increase mortality in patients supported with ECMO [76,84,85]. In patients treated with VA-ECMO, the prevalence of AIS is 3.3% and may be as high as 16% on autopsy [86,87]. The risk factors for AIS include circuit clots (oxygenator clot), left ventricle (LV) thrombus, and insertion of ECMO catheters [86,88-90]. The prevalence of ICH with VA-ECMO is reported to be between 2% and 18% and as high as 24% on autopsy [86,87]. Thrombocytopenia and heparin use pre-dispose to ICH and rapid hematoma expansion, which may be exacerbated by impaired coagulation function in ECMO patients [71,91,92].
Cerebral microbleeds (CMBs) were detected in 60% of patients treated with ECMO at autopsy and in 50% of survivors from a retrospective analysis, both of which are much higher than the rates in the general population [93,94]. Several other case series and retrospective observational studies also reported CMBs in patients treated with ECMO with unknown clinical significance or long-term outcomes [95-97]. CMBs may indicate ongoing cerebral small vessel disease in patients treated with ECMO, but their significance and etiology remain to be elucidated.
Harlequin syndrome
Harlequin syndrome, also described as the North-south syndrome, dual circulation, and differential hypoxia, is a phenomenon that occurs in peripherally cannulated VA-ECMO patients with respiratory failure and cardiac failure, such that they are unable to adequately oxygenate blood. The ECMO venous catheter drains oxygen from the inferior vena cava and returns oxygenated blood from the ECMO circuit to the aorta, which meets with the blood ejected from the LV that is recovering. This blood preferentially circulates towards the lower extremities and returns via the inferior vena cava, where it is recirculated through the ECMO circuit. Deoxygenated blood, due to pulmonary dysfunction, ejected from the LV may preferentially perfuse the aortic arch in an antegrade fashion, thereby perfusing the head vessels with deoxygenated blood as it competes with the oxygenated retrograde arterial blood flow from the femoral artery cannula. This mismatch of upper body hypoxia coupled with lower body normoxia, hence differential hypoxia, is called Harlequin syndrome [98-100]. Hypoperfusion and ischemia of the brain and heart are associated with HIBIs. The prevalence is reported to be approximately 9%; therefore, it is critical to monitor arterial blood gas values from the patient’s right radial artery, the most distal arterial access from the femoral artery cannula, to gain insights into the oxygenation status of the upper torso [101].
NEUROLOGIC MONITORING FOR ABI IN ECPR
Accurate estimation of the prevalence of ABI associated with ECPR is difficult due to limitations in neurological examination with the use of sedatives as well as safety concerns in performing neuroimaging studies during ECMO support. Magnetic resonance imaging is currently precluded because of the ECMO circuit [102]. Even if head computed tomography (HCT) is performed, the utility of HCT in detecting early ischemia and lesions in the posterior circulation territory is limited. Other noninvasive neurological monitoring methods include TCD, somatosensory evoked potentials, cerebral near-infrared spectroscopy (cNIRS), and electroencephalography (EEG), which may systematically assess the occurrence of ABI [103-105]. Another useful tool for noninvasive monitoring is optic nerve sheath diameter measurement with ultrasound, which may provide information on intracranial pressure [102]. A standard neurological monitoring protocol can increase sensitivity in the detection of ABI [103]. TCD may have a role in detecting ECMO circuit clots, such as arterial-sided oxygenator clots, which may be associated with ischemic stroke [89]. In addition, TCD can be used to detect ongoing cerebral microembolic signals while on ECMO, but further study is necessary to establish a firm relationship between AIS and TCD microembolic signals [106]. cNIRS may be a useful real-time bedside neuromonitoring tool to detect ABI in ECMO patients when an acute drop in regional oxygenation saturation occurs [107]. It is recommended to monitor patients after CA with continuous EEG (cEEG) [108]. Similarly, ECPR patients should be monitored with cEEG, as patients are at a high risk of seizures. Furthermore, EEG features such as absent EEG reactivity and discontinuous background may be associated with poor outcomes in comatose ECMO patients [109,110]. Therefore, cEEG monitoring is recommended for ECMO patients with disorders of consciousness off sedation and somatosensory evoked potentials for patients with motor Glasgow Coma Scale scores of < 4 [103,104]. Optic nerve sheath diameter can be abnormal due to ABI but is less useful in the prevention of injury [111]. Each of these neurological monitoring tools has limitations. Therefore, a standardized multimodal neuromonitoring approach, as well as clinical neurological assessment with neurological consultation, may facilitate early detection of ABI associated with ECPR. However, the effectiveness of this approach in improving outcomes by primary and secondary prevention or providing reliable neurological prognostic information is yet to be established [103]. Fig. 2 summarizes the neurological complications, monitoring, and prognostication of ECPR.
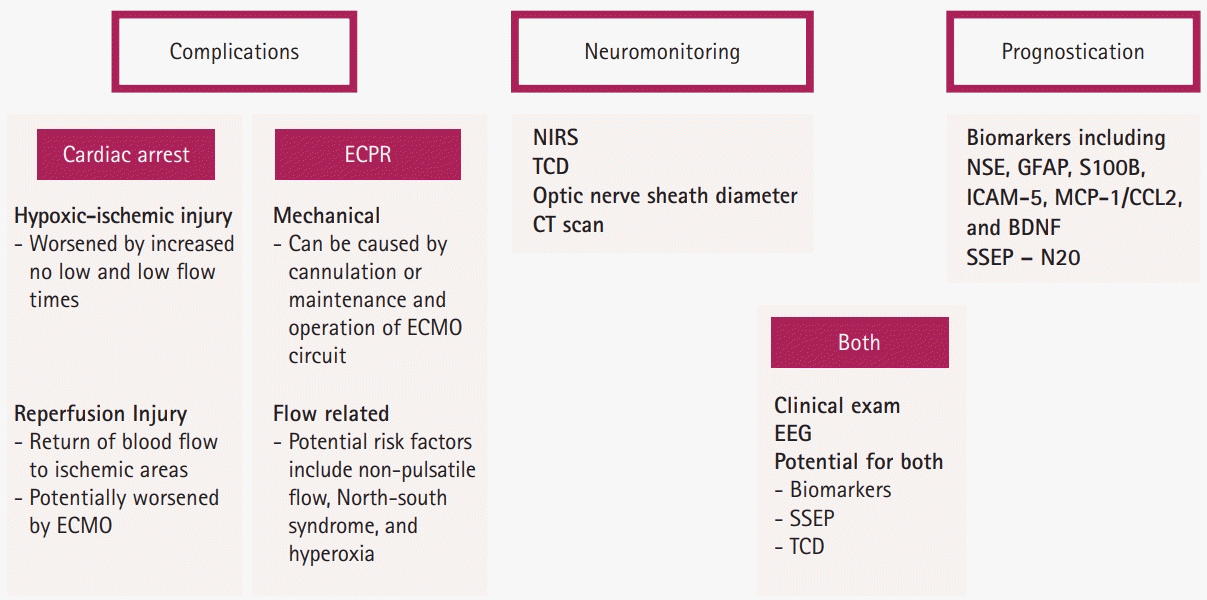
Components of neurologic complications, monitoring, and prognostication in extracorporeal cardiopulmonary resuscitation (ECPR). NIRS, near-infrared spectroscopy; TCD, transcranial Doppler; CT, computed tomography; NSE, neuron-specific enolase; GFAP, glial fibrillary acidic protein; S100B, calcium-binding protein B; ICAM-5, intercellular adhesion molecule 5; MCP-1/CCL2, monocyte chemoattractant protein 1/chemokine (C-C motif) ligand-2; BDNF, brain-derived neurotrophic factor; SSEP, somatosensory evoked potential; ECMO, extracorporeal membrane oxygenation; EEG, electroencephalography.
BIOMARKERS IN ECPR
Several biomarkers have been identified and associated with ABI in patients treated with ECMO. These include markers of neuronal injury (neuron-specific enolase [NSE] and intercellular adhesion molecule 5), glial cell injury (glial fibrillary acidic protein [GFAP], calcium-binding protein B [S100B], and brain-derived neurotrophic factor), and neuronal inflammation (monocyte chemoattractant protein 1/chemokine [C-C motif] ligand 2 [MCP-1/CCL2]) [102]. In patients treated with ECPR, higher NSE has been shown to correlate with increased mortality and ABI; however, hemolysis is common in ECMO and may result in false positives in NSE measurement [112,113]. In patients with ABI associated with ECMO treatment, S100B was found to be significantly elevated, and in a separate case series of infants, it was significantly elevated 72 hours before ICH [114,115]. There is evidence that GFAP is associated with ICH, brain death, cerebral edema, and mortality and is elevated in children 1–2 days before the detection of ABI on imaging [116]. S100B and GFAP in combination may be representative predictive biomarkers for children as the levels were elevated 1–3 days before the detection of ABI [115,116]. A marker of axonal injury in neurons, tau, measurable via serum assay may be significantly better than NSE for neuro prognostication after CA; however, there is a lack of data on the use of ECMO and EPCR [117]. Similarly, after CA, another marker of axonal injury, the neurofilament light chain, is associated with HIBIs in children and poor long-term neurologic outcomes in adults [118,119]. Post-CA biomarkers have been shown to peak at different periods but they are not sensitive or specific enough to be independent prognostic markers [120]. For ECPR, biomarkers may aid in prognostication as part of the multimodal evaluation, including imaging and clinical assessments, to increase sensitivity.
CONCLUSIONS
ECPR represents advancement in CPR, allowing a bridge to therapy in appropriately selected patients after refractory CA. Currently, ECPR is most successful at centers with experienced staff or communities with an appropriately trained and experienced ECMO team. Although ECPR may improve survival, ABI remains the leading cause of morbidity and mortality among patients treated with ECPR. A standardized neuromonitoring protocol may improve ABI detection. A better understanding of the role of early hyperoxia, TTM, cerebral blood flow, and reperfusion injury is important for improving neurological outcomes of ECPR survivors. Furthermore, cerebral microcirculation and autoregulation with non-physiological blood flow in ECMO may play a critical role in cerebral small vessel disease. “Bench-to-Bedside” translational and clinical research on “ABI in ECMO” is necessary as the use of ECPR is increasing, as well as its associated increase in survival.
Notes
Ethics statement
Not applicable.
Conflict of interest
No potential conflict of interest relevant to this article.
Acknowledgments
The authors would like to thank to the Departments of Neurology, Neurosurgery, Anesthesiology, and Cardiothoracic Surgery of Johns Hopkins Hospital.
Author contributions
Conceptualization: SMC, CJW. Data curation: NA. Formal analysis: NA. Funding acquisition: NA. Methodology: SMC, CJW. Project administration: SMC. Visualization: SMC, CJW. Writing–original draft: SMC, CJW. Writing–review & editing: SMC, CJW, CWC.