Management of propofol-related infusion syndrome and discussion of POLG mitochondrial mutation: a case report
Article information

Abstract
Background
Propofol-related infusion syndrome (PRIS) is a known complication of long-term propofol infusion. Providers should be aware of PRIS risk, as early recognition is key to avoiding mortality, which can range from 30% to 60%. The underlying mechanism of PRIS is unknown, but some studies suggest that underlying mitochondrial dysfunction may predispose patients to developing PRIS.
Case Report
We present a case of refractory adult-onset epilepsy that was challenging due to a paradoxical response to propofol with worsening brief ictal/interictal rhythmic discharges and complicated by development of PRIS. We aimed to discuss the clinical presentations of PRIS, along with a review of the mitochondrial POLG mutation found in our patient, which has also been described in other case reports of refractory adult-onset epilepsy.
Conclusion
We discuss the treatment strategy utilized in hopes of raising awareness of the risks in managing patients with epilepsy who have a potential underlying mitochondrial disorder.
INTRODUCTION
Propofol-related infusion syndrome (PRIS) is a known complication of long-term propofol infusion. Propofol was placed on the World Health Organization’s list of essential medicine in 2019, and has been used in many anesthetic settings since its discovery in 1977. Following propofol approval in 1989, the first report on PRIS was released in 1990, with the syndrome defined in 1989 by Bray et al. as a disease characterized by high anion-gap metabolic acidosis, rhabdomyolysis, hepatomegaly, lipemia, bradycardia, and eventual cardiovascular collapse that develops in patients on high-dose propofol infusions greater than 4 mg/kg/hr for more than 48 hours [1]. Providers should be aware of PRIS risk as early recognition is key to avoiding mortality, which can range from 30% to 60%. The underlying mechanism of PRIS is still unknown, but some studies suggest that an underlying mitochondrial dysfunction may predispose some patients to developing PRIS [2]. Mitochondrial mutations have been fully described in the literature with regard to their presentation in patients with pediatric epilepsy, but the genetics are poorly described in adult populations, with the two most common epileptic pathologies being mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes and myoclonic epilepsy with ragged red fibers [3]. When treating epilepsy with a potentially underlying mitochondrial defect, consideration should be made with regard to antiseizure drug (ASD) choice as some medications can theoretically compromise mitochondrial function [3]. We present a case where PRIS was successfully managed with identification of a possible underlying mitochondrial mutation.
CASE REPORT
Our patient was a 42-year-old African-American male with newly diagnosed right temporal lobe epilepsy who presented with status epilepticus and was subsequently intubated for hypoxia post-event. Upon admission, seizures were initially controlled with valproic acid and lacosamide. After 72 hours, the patient developed intermittent periods of agitation and desaturation. The patient’s electroencephalogram (EEG) became more active with new multifocal, independent sharp waves in the left frontal temporal region. Prior to admission, all electrographic spikes were observed only in the right temporal area. EEG activity continued to increase and evolved from brief ictal/interictal rhythmic discharges (BIRDs) to short bursts of seizure activity despite maximizing valproic acid and lacosamide doses and addition of fosphenytoin. The decision was made to place the patient in burst suppression, which was performed using fentanyl, midazolam, and propofol. Despite high doses of these medications, the patient’s EEG demonstrated an increased frequency of BIRDs, with discharges becoming more sharply contoured (Fig. 1). Ketamine was added, following which burst suppression was achieved.
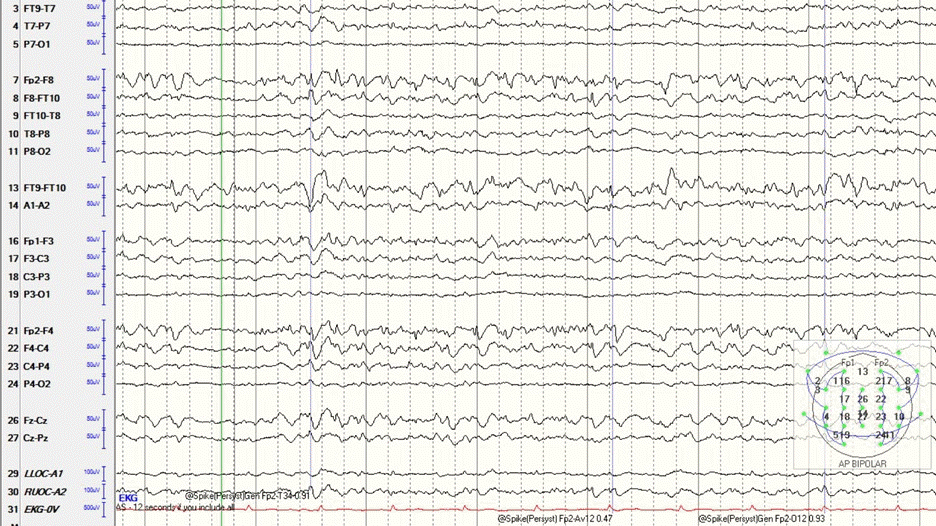
Electroencephalogram demonstrating the refractory brief ictal/interictal rhythmic discharges pattern.
After 48 hours, the patient developed new acidosis, elevated lactate, and elevated creatine phosphokinase levels. Within a few hours, he developed significant hypotension requiring pressors, and a new T-wave inversion was noted on electrocardiogram (Fig. 2). A medical workup including inflammatory and cardiac markers and cultures was conducted to rule out other causes such as septic shock. Treatment was escalated to broad-spectrum antibiotics, and a transthoracic echocardiogram was obtained, which demonstrated stable contractility. Propofol was infused at 4.5 mg/kg/hr for almost 72 hours until it was discontinued owing to clinical suspicion of PRIS. Despite immediate cessation of propofol, the patient’s bloodwork continued to demonstrate a rapid increase in creatine phosphokinase, triglycerides, lactate, and liver enzymes, with escalating vasopressor requirements. The nephrology department was urgently consulted, and the patient initiated continuous renal replacement therapy in the evening. Supplemental L-carnitine was added, valproic acid was discontinued, and the patient was transitioned to phenobarbital.

Electrocardiogram changes showing deep T wave inversions after lab abnormalities were discovered that were not present previously.
After propofol was discontinued, the burst suppression pattern improved, further ASDs decreased, and the previous BIRDs findings resolved and evolved into intermittent interictal spikes on EEG monitoring. The patient continued to stabilize over the next few days, and laboratory abnormalities resolved with continuous renal replacement therapy. Magnetic resonance imaging of the brain with and without contrast, along with arterial and venous phases, showed no structural or vascular abnormalities. A lumbar puncture combined with tests for abnormal epileptic mutations and autoimmune antibodies also revealed normalizing results. Genetic testing was performed through Mayo Laboratories combined mitochondrial analysis [4], and the patient tested positive for a likely pathogenic POLG gene mutation (c.3104+1G>A, Chr15:89862458) as well as two variants of uncertain significance, namely NDUFS8 (p.P7T) and TRMU (p.I99T) mutations. The patient’s mentation continued to slowly improve, and he was transferred to rehabilitation, where he was eventually discharged after a few weeks with his condition close to baseline on a regimen of phenobarbital, zonisamide, and lacosamide. He has since continued to experience a gradual decline in mentation with worsening memory loss and return visits for breakthrough seizures due to medication nonadherence.
DISCUSSION
It has been suggested that the pathophysiology of PRIS may be related to mitochondrial impairment through animal models and autopsies [5]. Recent literature is also supportive of this theory; several cases of PRIS have found that patients tended to have an underlying mutation affecting their mitochondria [2]. We describe a case of PRIS which was successfully managed with supportive care and early detection along with a genetic workup which included a genetic mitochondrial panel.
The POLG gene mutation in our patient can be seen in mitochondrial depletion syndromes, ataxia, and valproic acid-induced hepatotoxicity, which has also been described in other patients with PRIS [2]. Genetically, the POLG gene is known to encode mitochondrial DNA polymerase. Given the importance of mitochondria in aerobic metabolism and ATP generation, prior studies have proven its importance, with knockout mutations being embryonic lethal, and well over 200 mutations have been known to affect its function [6]. Disease severity can range from refractory infantile epilepsy to adult-onset ataxia. Because POLG mutations can present with vast clinical variations, it is important to note that apart from neurological involvement, there can also be effects on the cardiac, hepatic, gastric, and respiratory systems. In terms of neurological involvement, most described syndromes involve visual acuity, eye movements, hearing, epilepsy, ataxia, myopathies, and variable magnetic resonance imaging findings [6]. While it has been reported in small case series where there was a 23.1% prevalence of epilepsy with multifocal EEG abnormalities, this only made up 1.6% of the total number of patients with mitochondrial mutations that experienced epilepsy [3]. Further review of the POLG mutation and epilepsy demonstrates the potential for epilepsy refractory to medications along with subcortical myoclonus [7]. Our patient also tested positive for two variants of uncertain significance which included a NDUFS8 mutation that can be seen in autosomal recessive Leigh syndrome due to an impaired Complex I deficiency, and a TRMU mutation that can be seen in infantile liver failure.
Due to the paucity of evidence on mitochondrial mutations in adult-onset epilepsy, there are no guidelines regarding the selection of ASD. Among the three major medications used in status epilepticus, levetiracetam and fosphenytoin are not known to cause mitochondrial toxicity. Valproic acid should be used with care as some studies have reported potential toxicity, although there are also conflicting reports where patients with mitochondrial mutations tolerated the medication [5]. Patients with the POLG mutation developed refractory seizures, but phenobarbital and midazolam along with a ketogenic diet, a low glycemic diet, and a sodium channel blocker considered reasonable for first line therapy resulted in the best response [7]. Unlike other mitochondrial diseases, these patients have not been reported to have a baseline hepatopathy or myopathy, but the disease can be associated with valproate induced hepatotoxicity for which there have reports of successful rescue therapy with L-carnitine or N-acetylcysteine [7].
As the body of literature grows, it is possible that we may be able to identify a group of genetic mutations which place certain adult-onset epileptic patients at a higher risk of developing PRIS. Owing to the high mortality rate, it is important to be aware of PRIS as a deadly complication of high-dose propofol use. This report aims to provide further insight into the necessity of genetic testing in these patients, which may help identify a common link in the future. While it may not be feasible to screen all patients with mitochondrial disorders, patients with a known history should be cautioned about this catastrophic complication of a routine sedative, and patients who experience PRIS should undergo a mitochondrial workup to better characterize this disease and prevent future risks.
Notes
Ethics statement
Approval for this study was waived in accordance with UT Southwestern policies because this study was a case report of a single patient (no more than three patients) and did not include protected health information, data analysis, or testing of a hypothesis; and was de-identified. The requirement for informed consent was waived.
Conflict of interest
No potential conflict of interest relevant to this article.
Author contributions
Conceptualization: BN. Data curation: BN. Formal analysis: BN. Methodology: BN. Project administration: SF. Visualization: BN. Writing–original draft: BN. Writing–review & editing: all authors.